▎药明康德内容团队编辑
根据PDUFA的预期目标日期,预计10月,美国FDA将对1个创新药物的批准做出监管决定。如果审批流程速度较快,另有3个创新药物有希望在10月迎来审批决定。
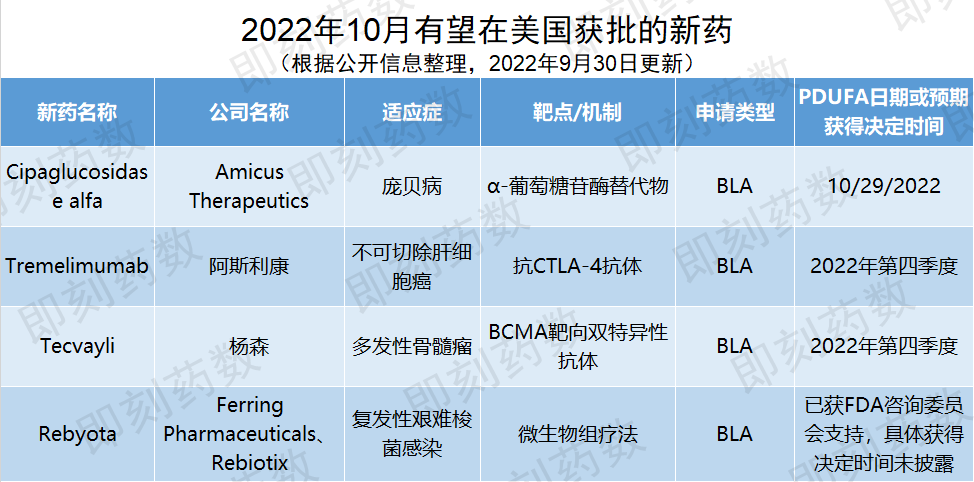
▲10月有望在美国获批的新药(药明康德内容团队制图,点击可见大图)
药物名称:Cipaglucosidasealfa(疗法AT-GAA中的组分)
公司名称:AmicusTherapeutics
适应症:庞贝病

Amicus Therapeutics的组合疗法AT-GAA(cipaglucosidase alfa+miglustat)是治疗庞贝病的酶替代疗法(ERT)。Cipaglucosidase alfa是一种α-葡萄糖苷酶替代物,而另一个组分miglustat则是一种口服酶稳定剂,旨在增强cipaglucosidase alfa的活性。庞贝病(糖原累积病II型)是因α-1,4-葡萄糖苷酶遗传缺陷导致,进而引发糖原堆积在溶酶体和胞质中,造成心肌、骨骼肌等一系列脏器损害的罕见病,发病率约为1/4万~1/5万。
FDA已经接收了cipaglucosidase alfa的生物制品许可申请(BLA),PDUFA日期设定为2022年10月29日。此前,FDA已授予AT-GAA孤儿药资格以及突破性疗法认定。3期临床结果显示,AT-GAA相比标准治疗方式alglucosidase alfa,在治疗迟发性庞贝病患者(包括已经过ERT治疗但治疗需求尚未得到满足的患者)时获得具有临床意义的改善效果。如果获得批准,该疗法可能成为治疗庞贝病的新治疗标准。
药物名称:Tremelimumab
公司名称:阿斯利康
适应症:不可切除肝细胞癌
阿斯利康(AstraZeneca)的tremelimumab是一种人源化的抗CTLA-4抗体,可以阻断CTLA-4、促进T细胞活化并加强对癌症的免疫反应。目前,该药物正在寻求美国FDA批准,与抗PD-L1抗体Imfinzi(durvalumab)联用,治疗不可切除的肝细胞癌患者。这一称为STRIDE的创新给药方案首先使用单剂tremelimumab治疗,然后患者持续接受Imfinzi治疗。肝癌是世界上第六大常见癌症,每年有大约90万患者确诊。它同时是导致癌症死亡的第三大原因,大约只有7%的晚期患者能够生存超过5年。
FDA于4月25日接收了该药物的BLA并授予其
优先审评资格
,PDUFA日期设定为2022年第四季度,按照一般优先审评程序所需的6个月时间估计,tremelimumab有可能于10月迎来审批决定。这一BLA是基于3期临床试验HIMALAYA的最终结果。在这一试验中,接受STRIDE给药方案治疗的患者的死亡风险与活性对照组相比降低22%(HR=0.78,96.02% CI,0.65-0.93,p=0.0035)。接近三分之一(31%)的患者生存期超过3年,而活性对照组这一数值为20%。如果获得批准,该疗法将为晚期肝癌患者提供新的治疗方案选择,从而改善患者的长期生存结果。
药物名称:Tecvayli
公司名称:杨森
适应症:多发性骨髓瘤

强生(Johnson& Johnson)旗下杨森(Janssen)的Tecvayli是一款靶向B细胞成熟抗原(BCMA)的双特异性抗体,拟用于治疗复发/难治性多发性骨髓瘤(RRMM)成人患者。Tecvayli是通过将T细胞募集到表达BCMA的骨髓瘤细胞附近,激发T细胞杀伤肿瘤细胞的潜在“first-in-class”双特异性抗体疗法,已获得FDA授予的孤儿药资格以及突破性疗法认定。多发性骨髓瘤是血液系统的第二常见恶性肿瘤,约占血液系统恶性肿瘤的10%。目前它仍然是一种无法治愈的血液癌症,几乎所有患者会出现复发并且需要接受后续治疗。
1/2期临床试验结果显示,在165名接受每周皮下注射Tecvayli的RRMM患者中,总缓解率达到63%,值得一提的是,58.8%的患者获得非常好的部分缓解(VGPR)以上的应答,39.4%的患者获得完全缓解(CR)以上的应答。此外,患者的中位缓解持续时间达到18.4个月,中位无进展生存期为11.3个月,中位总生存期为18.3个月。该药物的BLA已于2021年12月递交至FDA,有望于今年第四季度迎来审批决定。如果近期获批,Tecvayli将成为美国首款获批治疗多发性骨髓瘤的双特异性疗法,也是首款获批靶向BCMA的双特异性抗体,为难治性患者提供了一种“现货型”治疗手段。
药物名称:Rebyota
公司名称:FerringPharmaceuticals、Rebiotix
适应症:艰难梭菌感染
Ferring Pharmaceuticals和旗下Rebiotix共同研发的Rebyota是预防艰难梭菌感染(CDI)的微生物组疗法,它不含任何抗生素,旨在帮助患者恢复肠道微生物群落。该疗法已被美国FDA授予快速通道资格、孤儿药资格和突破性疗法认定。CDI是一种衰弱性疾病,可以造成严重的腹泻、发烧、胃痛、食欲下降、恶心、盲肠感染,显著地影响患者的生活质量。据预估,约35%的CDI确诊者会发生复发,而在经历过第一次CDI复发的患者中,有65%的病患会再次产生复发。
9月24日,美国FDA的疫苗与相关生物制品产品咨询委员会(VRBPAC)成员以13:4的投票
结果
,认为Rebyota的临床数据足以支持其所宣称疗效,即可有效减少在18岁以上复发性CDI成人病患在接受抗生素治疗后的感染复发。此外,咨询委员会亦以12:4的票数(1票弃权)支持Rebyota治疗的安全性。FDA在审评药物生物制品许可申请(BLA)时不须遵从咨询委员会的决定,但这一结果无疑增加了Rebyota成为首个获批的微生物组疗法的可能性。