“每一个孩子出生时都带来了讯息—上帝对人类并未灰心失望。”—泰戈尔
据说每个孩子都是上帝派来人间的礼物,但却有这样一些小“天使”,他们出生时外表和其他宝贝一样,脸上却始终洋溢着灿烂的笑容,殊不知,是被命运开了玩笑......
你的笑容,令人心疼
1965年,英国儿科医生Harry Angelman首次报道了3个智力低下、总是微笑、不会说话的小病人,神似Castelvecchio博物馆一幅油画中一个孩子完成木偶以后得意的笑容,故称之为“快乐木偶病”。
后续研究发现,这是一种罕见的、遗传性、神经发育异常综合征,同时为了纪念,“天使综合征(Angelmansyndrome,AS)”由此得名。

“天使综合征”患儿[1]油画中的笑容
一.发病机制
AS是由母源15q11-13染色体区域的UBE3A基因表达异常或功能缺陷导致的神经发育障碍性疾病。在欧美人群中的患病率为1/24,000~1/12,000,我国多为散发,尚无相关流行病学调查报告。
患者的神经系统,主要是黑质、纹状体、海马及小脑浦肯野细胞的泛素化异常,在胎儿期、新生儿期的临床表型多为正常,通常于1岁以后出现典型的AS症状[2]。
二. 临床表现
参照2005年版临床诊断标准共识,AS的临床特征依发生频率可分为[3,4]:

1、均出现的表现(100%患者)
(1)发育迟缓:发育里程碑延迟而无倒退;
(2)运动或平衡障碍:通常为共济失调和/或肢体震颤;
(3)语言障碍:无或极少量词汇,重复性语言和非语言交往能力强于表达性语言能力;
(4)异常行为特征:频繁大笑或微笑、明显的兴奋动作或快乐举止,常伴拍手或多动;
(5)正常孕产史和出生头围,无出生缺陷和生化指标异常。
2、经常性表现(>80%患者)
(1)头围小或增长缓慢(
(2)癫痫发作(3岁前);
(3)异常脑电图:特征性高波幅棘-慢波。
3、相关性表现(20%~80%患者)
枕部扁平、喜吐舌、流涎、婴儿期喂养困难、肌张力低、过度咀嚼动作、斜视、嘴大牙缝宽、与家人相比皮肤色素减退(仅见于缺失型)、下肢腱反射亢进、行走时喜上肢抬起、热敏感性增加、步基宽、脚踝内翻或外翻、睡眠差、肥胖(多见于年长、非缺失型患者)、脊柱侧弯、便秘等。

AS的临床特征[4]
三. 辅助检查
1、遗传学检查
为该病确诊手段。主要包括DNA甲基化分析(甲基化多重连接依赖式探针扩增技术/MS-MLPA、甲基化PCR)和UBE3A基因序列分析。此外,SNP-array芯片分析可以检测15号染色体的微缺失和父源单亲二体。
2、脑电图
在患者出现明显的临床症状或基因确诊前,有助于此病的早期诊断。目前公认的AS特征性脑电图表现有:①δ图形;②θ图形;③后头部棘慢波。
四.诊断
符合AS临床诊断标准共识和(或)分子遗传学检测结果表明母源UBE3A等位基因存在表达或功能缺陷时,即可给予诊断。
不同发病机制患者在诊断过程中采取的诊断手段不同。遗传学检测首选DNA甲基化分析,可诊断母源15q11.2-q13缺失、父源单亲二体或印记缺陷所致的AS,约占所有患者的80%。
DNA甲基化分析正常者可进行UBE3A基因序列分析,该方法可使大约10%的AS患者得到确诊。另外,仍有约10%患者由于尚未明确的致病机制而无法实现分子遗传学确诊,只能依据典型表现做出临床诊断。
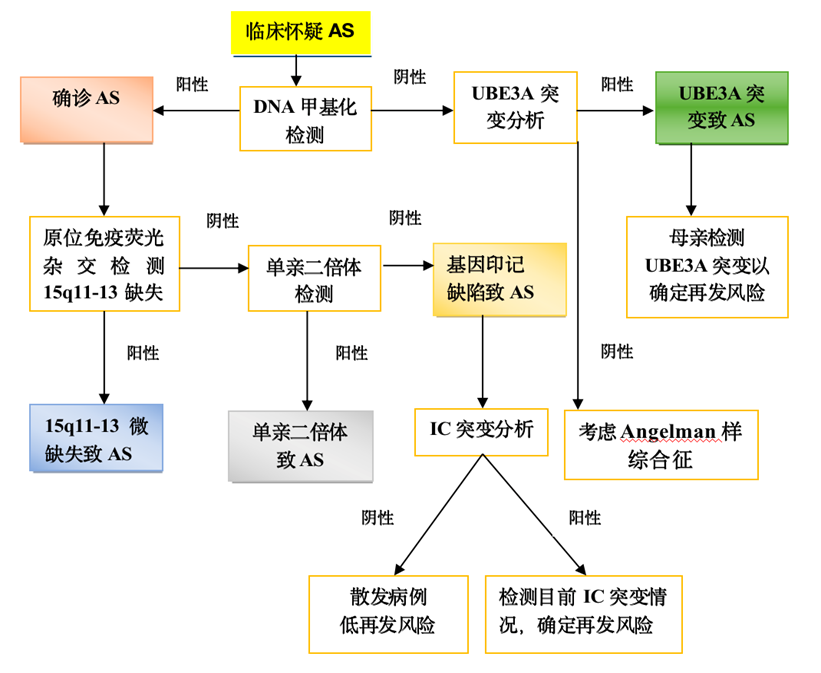
AS诊疗流程图[2]
五. 治疗
AS迄今尚无特效治疗,主要工作在于诊断后健康管理。目前主要是针对喂养困难、癫痫、行为异常、言语障碍、脊柱侧凸等临床表现进行积极的对症及支持治疗,以提高患儿的生活质量。
分子靶向疗法是当下AS治疗的研究热点,也是唯一可能的治愈方法。
一些基因治疗正在积极开展中,比如利用端粒酶抑制剂或反义寡核苷酸激活甲基化沉默的父源UBE3A等位基因。以期达到增加父源UBE3A等位基因的表达、补充母源UBE3A等位基因缺陷、最终治疗疾病的目的。